UK Updates- MHRA Consultation on its Fee Proposal for 2025.

Consultation on proposals to update the MHRA's statutory fees to ensure they continue to recover their costs.
The Medicines and Healthcare products Regulatory Agency (MHRA) is seeking views from interested stakeholders on proposals to update its statutory fees to ensure they continue to recover their costs.
The MHRA’s fees are updated on a regular basis to ensure they continue to achieve full cost-recovery in line with HM Treasury guidance ‘Managing Public Money’. This is necessary for long-term financial sustainability and the ongoing delivery of the MHRA’s services. The implementation date for these proposed changes is April 2025.
Information Sought
The Medicines and Healthcare products Regulatory Agency (MHRA) is seeking views on the following proposals:
Increasing the statutory fees shown in its consultation document to ensure continued cost-recovery until 2027. This cost recovery covers for services such as:
Recovered Cost includeAmending the existing Medical Device Registration fee to include the costs for medical device post-market work.
Creating a new service providing regulatory advice meetings for medical devices.
Amending the fee model for three existing services, as well as increasing the fees to ensure continued cost-recovery until 2027, and also to remove 51 fees that are no longer in use.
Updating and clarifying the legal definition of a “standard variation” application for homeopathic products. The “standard variation” application statutory fee for homeopathic products will be unchanged.
If you have any questions about your consultation response or are experiencing any problems, please email info@sushvin.com The consultation document can be found here: https://www.gov.uk/government/consultations/mhra-consultation-on-statutory-fees-proposals-on-ongoing-cost-recovery
Timing The MHRA is aiming to implement the new fee structure in April 2025.
Application of New Fee New fees would apply from April 2025 to March 2027. This allows fees review every two years.
Provide Your Feedback The survey will run from 29 August 2024 and closes at 11:59pm on 24 October 2024. The link to participate is: HERE
UK MHRA Fee Increase: Open Consultation - Implications for Manufacturers of Medical Devices
The MHRA is considering two fee changes for medical devices, under Proposal 2 and Proposal 3: HERE
Proposal 2 – Annual Registration Fee
Currently, the MHRA only charges a fee during the initial device registration process. However, they are now proposing to collect an annual registration maintenance fee, to help cover their ongoing post-market activities. The suggested fee is £210 per GMDN code.
The MHRA included a table assessing the impact across all registered devices. A large number of companies are shown to have only one GMDN code registered. However, the cost can become significant for companies with a high volume of devices. For example, if a manufacturer has 100 GMDN codes registered, they can expect to pay £21,000 per year.
How will fees be collected? The proposal states:“It is intended that the MHRA registration system, with some modification, will automate the initial registration charge and its annual renewal.”
Proposal 3 – Regulatory Advice Meetings
The MHRA intends to launch a new service providing “expert regulatory advice to medical device manufacturers”. It is intended particularly for “novel and/or complex products with the potential to significantly improve patient outcomes, where the application of the Regulations is not straightforward and easily understood”.
The MHRA acknowledges that they already provide this type of support at no cost; however, charging a fee would allow more resources to be made available. The proposed fee is: “… £987 for a one hour meeting. This is based on the cost of internal MHRA discussions, meeting preparation, the meeting and any post meeting feedback.”
MHRA Fees – Proposals (In comparison to existing fees)
| Service Type | Percentage Increase/Fee |
|---|---|
| Clinical Amendment/Consultation | 9.1% |
| Clinical Notification | 104.9% |
| Drug-Device Combination | 8.9% |
| Approved Body Designation and Monitoring | 15.9% |
| Registration | £210 annual fee per GMDN code |
| New Regulatory Advice Meetings | £987 per hour |
| Source: Association of British HealthTech Industries (ABHI) | |
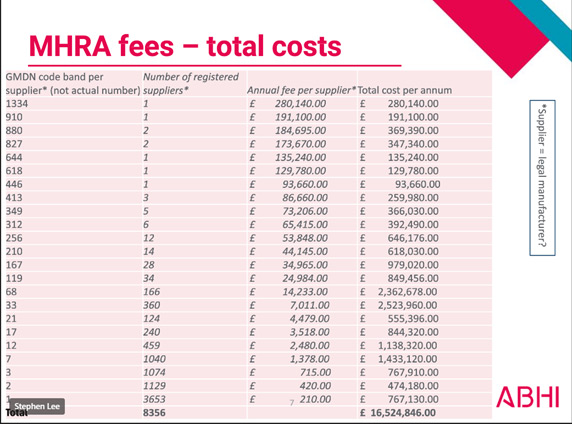
MHRA Fees – Total Costs
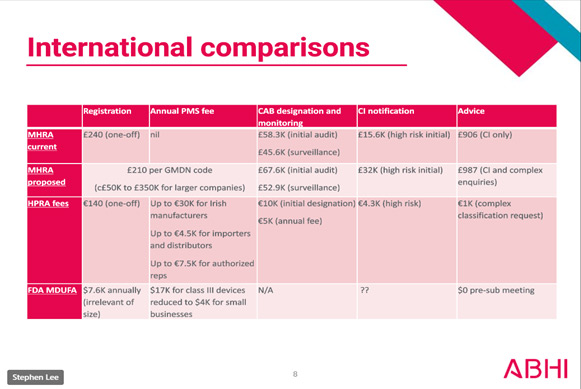
International Comparison
Cost of fee changes to Medical Device Manufacturers
The MHRA are proposing to update their registration. It is currently set at £240 per registration (not that long ago it was £100), but now they are proposing to increase it to £210 per GMDN code!
Take for an example, surgical instrument manufacturers often have more than 150 different GMDN codes applicable to their product range - this would mean an increase in registration cost to £31,500 - a 15,000% cost increase! Registration is not a one-off cost either; they are proposing to make it an annual fee. Registration in EUDAMED is currently free! In comparison to other international health authorities, MHRA’s proposed fee changes are 5-10X to that of USA’s FDA and Ireland’s HPRA. If you value sanity in our medical device regulations and don't want any more manufacturers pulling out of the UK market than have done already, please respond to their consultation here: HERE. Lower risk devices will be hit hardest if these changes go ahead, which is the opposite to how it should be. If the MHRA also go ahead with their plans to remove the "in-house manufacturing exemption" then multiple departments in each hospital will also have to register their devices too, which will result in fees running into tens of thousands for each NHS trust as well!

Sushvin Provides UK Responsible Person (UK RP) Services
Are you a legal medical device/IVD manufacturer located outside the UK and would like to market your product in the UK, then you will require a UK responsible person who will act on behalf of the manufacturer to ensure all the responsibilities detailed within the updated UK MDR 2002 regulations are met. As a legal manufacturer, organisations will have to register their devices prior to be placed on the UK market. If you need UK Responsible Person (UK RP) services, please contact SUSHVIN for more information.
Swissmedic, Switzerland Updates

For the inspectorates, to whom all technical interpretations are primarily addressed, the technical interpretation applies with immediate effect, as always in such cases. Numerous clarifications have been made in the new version. In most cases, compliance with the previous technical interpretation also guarantees compliance with the current version. In the few situations in which this is not the case, the licence holder is generally required to adapt immediately.
Chapter 5.5 requires a good knowledge of the local language for the responsible person. Establishment licence holders whose already approved responsible person does not meet these requirements are given a period of 12 months from the date of publication of the updated technical interpretation to make the necessary adjustments, either by appointing a new responsible person with the required language skills or by ensuring that the responsible person acquires the required language skills within this period.
Purpose and Scope
The requirements for a Responsible Person (RP) are described in art. 5, 6, 17, 18, 22, 23, 25, 26 and 39 of the Medicinal Products Licensing Ordinance (MPLO). This Technical Interpretation describes the interpretation of these articles by the Swiss inspectorates. It can also be used by companies as a basis for assessing whether an individual fulfils the requirements for applying to Swissmedic to act as a Responsible Person.
USFDA Updates

The flowchart shown below illustrates the MDSAP audit sequence and interrelationships.
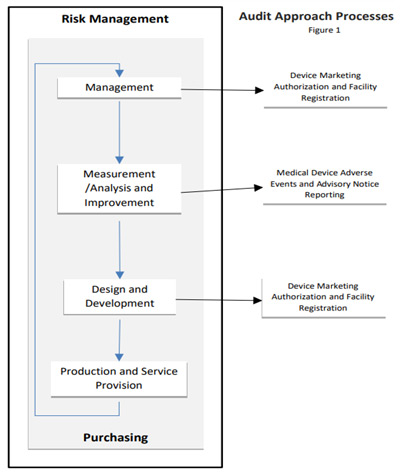
The design of the Medical Device Single Audit Program (MDSAP) audit process is to ensure a single audit will provide efficient yet thorough coverage of regulatory requirements. These requirements include; Medical devices – Quality management systems – Requirements for regulatory purposes (ISO 13485:2016), the Quality Management System requirements of the Conformity Assessment Procedures of the Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3), the Brazilian Good Manufacturing Practices (RDC ANVISA 665/2022), the Canadian Medical Devices Regulations, the Japanese Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No. 169), the Quality System Regulation (21 CFR Part 820), and specific requirements of the medical device regulatory authorities participating in the MDSAP program.
eMDR System Enhancements
FDA has published a page listing enhancements to CDRH's Electronic Medical Device Reporting (eMDR) system. https://www.fda.gov/medical-devices/emdr-electronic-medical-device-reporting/emdr-system-enhancements?utm_medium=email&utm_source=govdelivery
This page lists enhancements to CDRH's Electronic Medical Device Reporting (eMDR) system. The FDA eSubmitter client is updated concurrently with the eMDR system. Industry with system-to-system, or AS2, accounts with the FDA Electronic Submissions Gateway (ESG), should use the information on this page to plan system updates to align with these eMDR system enhancements as soon as possible.
The FDA recognizes the importance of providing early notice and predictability about potential eMDR system changes, especially for manufacturers submitting HL7 ICSR XML reports using AS2. Therefore, the FDA is implementing enhancements to the eMDR System on a regular schedule. Through this schedule, the FDA expects to take the next steps for every cycle of new enhancements:
- 1. Announce upcoming enhancements in June,
- 2. Release the Implementation Package in August,
- 3. Deploy enhancements to pre-production (ESG Test) in September, and
- 4. Deploy high-impact enhancements to production 7 months after the Implementation Package is released, typically in March of the following year.
While the FDA expects to follow this schedule for future enhancements, it is possible that emergency fixes may need to be implemented outside the schedule. Additionally, if major system changes are necessary in the future, additional time will be provided between release of the implementation package and production deployment.
FDA will provide regular communications on this page at each point in the process. This information will also be emailed to the CDRH Industry's email list. Please contact the eMDR helpdesk at eMDR@fda.hhs.gov for any questions or concerns about specifications and enhancements. Industry with AS2 accounts may use eMDR Test (through ESG Test) for testing system updates at any time.
FDA has published a Guidance Document on Acceptable Media for Electronic Product User Manuals - Docket Number: FDA-2020-D-0957
FDA is issuing this guidance to allow manufacturers to provide user manuals accompanying electronic products in either paper or electronic form. This is done to recognize that electronic media are now being widely used to provide instruction, while at the same time reducing paper consumption, increasing accessibility and providing rapid means for editing and updating content.
Australia Updates

TGA has recently updated CTA (Clinical Trial Approval) scheme forms All CTA scheme forms should be submitted to clinical.trials@health.gov.au (PDFs or word). Electronic/digital signatures can be used. If sending the forms by email (recommended), you are not required to send physical (hard-copy) copies to the mailing addresses detailed in each form. The CTA application contains two parts: the application form and supporting data.
Application form
Before commencing the trial, the sponsor must submit the application form. This form provides details of the sponsor and the ‘unapproved’ goods proposed to be used in the trial (medicine, biological and/or medical device).Supporting data
The sponsor of the trial should submit supporting data in an electronic dossier (searchable PDFs). This should be sent via email to clinical.trials@health.gov.au - if the file is too large, please contact TGA for advice.
Commencement of the trial and addition of trial sites
The sponsor must notify the TGA within 28 days of commencing supply of the goods at each site for each new trial conducted under the CTA scheme as well as additional sites in ongoing CTA trials.
Completion of a trial
The sponsor should notify the TGA of the completion of trials conducted under the CTA scheme after the trial has been completed at all sites. This would usually correspond with the last patient’s last visit. It is not necessary to notify completion dates for individual trial sites.
Health Sciences Authority (HSA), Singapore Updates

The Medical Devices Cluster (MDC) has released a draft of Guidance on Change Management Program (CMP) for SaMD for stakeholders' consultation: August 2024 Software as a Medical Device (SaMD), including those incorporating Machine Learning (ML) technology, plays a crucial role in offering innovative solutions to improve medical diagnosis, treatment, and patient care. SaMD Product Owners (PO) are required to adopt Total Product Life Cycle (TPLC) approach to manage and adapt to the ever-changing SaMD while ensuring that the software remains relevant, safe and effective throughout its life cycle.
However, prevailing regulatory framework may not be suited to accommodate the rapid iterative nature of SaMD. POs face the challenges of adhering to regulatory requirements which consist of ensuring compliance and obtaining regulatory approvals which can impact the timeliness of the implementation of SaMD changes. Hence, adoption of modern regulatory framework that embraces agile methodologies and risk-based assessments is necessary to help to expedite the approval process for certain types of changes, especially those aimed at improving the effectiveness and safety of SaMD.
To address this, the Health Sciences Authority (HSA) has initiated a new optional regulatory pathway – Change Management Program (CMP), specifically for SaMD that is incorporated into HSA’s Premarket Product Registration and Change Notification (CN) processes. The document describes the regulatory requirements and procedures for CMP submission. CMP is a new optional regulatory pathway specifically for SaMD that is incorporated into HSA's Premarket Product Registration and Change Notification (CN) processes, which also introduces the concept of Pre-specified changes. https://www.hsa.gov.sg/announcements/regulatory-updates/consultation-on-guidance-on-change-management-program-(cmp)-for-samd The Health Sciences Authority (HSA) would like to invite their stakeholders to provide feedback on the Guidance on Change Management Program (CMP) for SaMD The Consultation period for this document is from 26 August 2024 to 21 October 2024. Please email your feedback using the prescribed feedback form to HSA_MD_INFO@hsa.gov.sg by 21 October 2024 and with "Feedback on Guidance on CMP for SaMD" in the email subject header.
FAQ's
What is MHRA consultation on statutory fees about?
The aim of this consultation is to seek views on proposals to update the statutory fees charged by the MHRA to ensure it continues to recover its costs. The consultation will run from 29 August 2024 and close on 24 October 2024. The MHRA’s fees are updated on a regular basis to ensure they continue to achieve full cost-recovery in line with HM Treasury guidance “Managing Public Money”. This is necessary for long-term financial sustainability and the ongoing delivery of the MHRA’s services. The implementation date for these proposed changes is April 2025.
What is new in the newly published version of the MDSAP AUDIT APPROACH Document No.: MDSAP AU P0002.009?
This revision of the document combines the formerly separate MDSAP Audit Model and Process Companion documents into a single document containing additional detail regarding each audited process; as well as guidance for assessing the conformity of each process. In electronic form, the navigation bar facilitates quick access to relevant Tasks. The user may create their own bookmarks to quickly navigate to various sections.
What is the regular schedule of the FDA for implementing enhancements to the Electronic Medical Device Reporting (eMDR) System?
The FDA recognizes the importance of providing early notice and predictability about potential eMDR system changes, especially for manufacturers submitting HL7 ICSR XML reports using AS2. Therefore, the FDA is implementing enhancements to the eMDR System on a regular schedule. Through this schedule, the FDA expects to take the next steps for every cycle of new enhancements-
1. Announce upcoming enhancements in June 2. Release the Implementation Package in August 3. Deploy enhancements to pre-production (ESG Test) in September and 4. Deploy high-impact enhancements to production 7 months after the ImplementationPackage is released, typically in March of the following year. While the FDA expects to follow this schedule for future enhancements, it is possible that emergency fixes may need to be implemented outside the schedule. Additionally, if major system changes are necessary in the future, additional time will be provided between release of the implementation package and production deployment. The FDA will provide regular communications on eMDR System Enhancements page at each point in the process. This information will also be emailed to the CDRH Industry's email list. Please contact the eMDR helpdesk at eMDR@fda.hhs.gov for any questions or concerns about specifications and enhancements. Industry with AS2 accounts may use eMDR Test (through ESG Test) for testing system updates at any time.
Does the FDA have a Guidance Document on Electronic Media for Electronic Product User Manuals?
FDA has recently published a Guidance Document on Acceptable Media for Electronic Product User Manuals - Docket Number: FDA-2020-D-0957 FDA is issuing this guidance to allow manufacturers to provide user manuals accompanying electronic products in either paper or electronic form. This is done to recognize that electronic media are now being widely used to provide instruction, while at the same time reducing paper consumption, increasing accessibility and providing rapid means for editing and updating content.
Where should the TGA’s CTA (Clinical Trial Approval) scheme forms be submitted?
All CTA scheme forms should be submitted to clinical.trials@health.gov.au (PDFs or word). Electronic/digital signatures can be used. If sending the forms by email (recommended), you are not required to send physical (hard-copy) copies to the mailing addresses detailed in each form.










